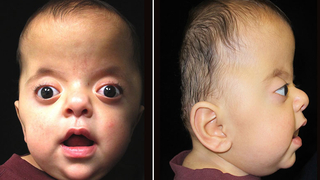
Síndrome de Goldenhar
Síndrome de Goldenhar, también conocido como el espectro óculo-aurículo-vertebral
El Síndrome de Goldenhar constituye una anomalía craneofacial compleja originada por una interrupción en la migración de las células de la cresta neural, lo que afecta directamente el desarrollo del primer y segundo arco branquial durante el periodo crítico de las semanas cuatro a ocho de gestación.
Aunque históricamente se ha considerado una condición de aparición esporádica, los avances genéticos han identificado variantes en el gen SF3B2 como responsables de […]