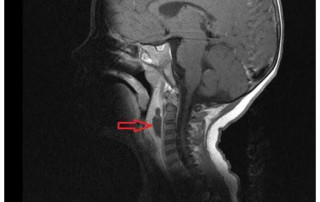
Absceso periamigdalino y retrofaríngeo
Las infecciones de los espacios profundos del cuello representan un urgencia por la posibilidad de complicaciones graves como la sepsis y la obstrucción de la vía aérea.
El absceso periamigdalino y el absceso retrofaríngeo comparten la necesidad de una cobertura antibiótica de amplio espectro. Sin embargo, sus presentaciones clínicas, anatomía y estrategias de manejo tienen matices diferentes.
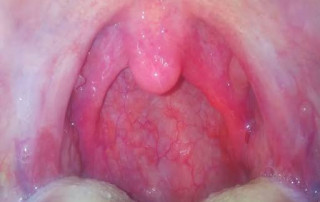
El absceso periamigdalino se manifiesta comúnmente con un dolor de garganta severo, fiebre y la […]